Concept article: Minireviews
Sequence Alignment & Algorithms in Bioinformatics
RACHEDI Abdelkrim
Laboratory of Biotoxicology, Pharmacognosy and biological valorisation of plants, Faculty of Sciences, Department of Biology, University of Saida - Dr Moulay Tahar, 20100 Saida, Algeria.
📧 E. mail: abdelkrim.rachedi@univ-saida.dz
Published: 15 February 2026
Abstract
Sequence alignment is a fundamental tool in bioinformatics used to compare and analyze biological sequences such as DNA, RNA, and proteins. This technique involves arranging two or more sequences to identify regions of similarity and difference, which may reflect functional, structural, or evolutionary relationships. Beyond its descriptive role, sequence alignment is inherently an algorithmic problem, where the objective is to determine an optimal or near-optimal alignment according to a defined scoring system. Over the years, a variety of computational algorithms have been developed to address this problem, ranging from exact methods based on dynamic programming, such as the Needleman–Wunsch algorithm and Smith–Waterman algorithm, to heuristic approaches like BLAST that enable rapid searches in large biological databases.
These algorithms rely on mathematical frameworks incorporating scoring schemes, substitution matrices, and gap penalties to evaluate alignment quality and biological plausibility. Metrics such as percentage identity and similarity, along with matrices like PAM and BLOSUM, allow for a quantitative interpretation of sequence relationships. In addition, modern bioinformatics tools integrate these algorithms into user-friendly platforms for large-scale analysis.
Sequence alignment plays a critical role in numerous applications, including the inference of evolutionary relationships, the identification of conserved functional regions, the detection of mutations associated with diseases, and the design of therapeutic strategies. This article presents a historical overview of sequence alignment, introduces its fundamental concepts, and explores the underlying algorithms, scoring methods, and computational tools, highlighting their importance in biology, biotechnology, mutation discovery, and disease research.
Key words
Primary structure, Sequence alignment, Algorithms, Bioinformatics, Phylogenetic trees, Mutations, Scoring Matrices, PAM, BLOSUM, Biological Function
Introduction
The advent of modern sequencing technologies has revolutionized biology by enabling the acquisition and analysis of genetic information at an unprecedented scale. High-throughput sequencing platforms now generate vast amounts of genomic and proteomic data; however, the mere determination of a nucleotide or amino acid sequence is insufficient to fully elucidate its biological function, structural properties, or evolutionary history. To extract meaningful insights, computational methods are required to compare sequences and interpret their relationships in a systematic and quantitative manner.
Sequence alignment has a long and foundational history in bioinformatics, dating back to the 1960s when Margaret Dayhoff introduced the concept of protein sequence comparison and developed one of the first protein sequence databases (Dayhoff et al., 1966). Her work led to the formulation of the PAM (Point Accepted Mutation) matrices, which provided a probabilistic framework for quantifying amino acid substitutions over evolutionary time (Durbin et al., 1998). These early developments established sequence alignment as both a biological and computational problem, laying the groundwork for the development of formal algorithms.
From an algorithmic perspective, sequence alignment can be formulated as an optimization problem in which the goal is to determine the best correspondence between two or more sequences according to a defined scoring system. This has led to the development of several classes of algorithms. Exact methods based on dynamic programming, such as the Needleman–Wunsch algorithm (Needleman & Wunsch, 1970) and the Smith–Waterman algorithm (Smith & Waterman, 1981), guarantee optimal solutions by systematically exploring all possible alignments. However, due to their computational complexity, heuristic methods such as BLAST (Altschul et al., 1990) and FASTA were developed to enable rapid searches in large biological databases by approximating optimal alignments. More advanced probabilistic approaches, including hidden Markov models, further extend alignment methodologies by incorporating statistical models of sequence evolution (Durbin et al., 1998).
Sequence alignment is thus a fundamental tool in bioinformatics used to compare and analyze DNA, RNA, and protein sequences. By aligning sequences to identify conserved and variable regions, researchers can infer evolutionary relationships, predict functional domains, detect mutations associated with diseases, and support drug discovery efforts. The evaluation of alignments relies on scoring schemes that integrate substitution matrices, such as PAM and BLOSUM, along with gap penalties to reflect biologically realistic evolutionary events.
There are two principal types of sequence alignment: pairwise alignment and multiple sequence alignment (MSA). Pairwise alignment focuses on comparing two sequences, whereas MSA extends this comparison to multiple sequences simultaneously, allowing for the identification of conserved motifs and the reconstruction of evolutionary relationships. Algorithms for MSA, such as progressive alignment methods implemented in tools like ClustalW, combine heuristic strategies with pairwise alignment principles to manage the increased computational complexity.
In practice, sequence alignment is implemented through a wide range of bioinformatics tools that integrate sophisticated algorithms into accessible platforms. Programs such as the Basic Local Alignment Search Tool - BLAST (Altschul et al., 1990) and ClustalW are widely used for sequence comparison and database searching. These tools enable applications across biology and biotechnology, including phylogenetic tree reconstruction, identification of disease-causing mutations, comparative genomics, protein structure and function analysis, and rational drug design.
The sequence alignment, thus, represents a cornerstone of modern bioinformatics, bridging biological data and computational algorithms. Its continued development reflects advances in both algorithm design and biological understanding, making it an indispensable approach for studying the structure, function, and evolution of biomolecules.
Methods
Algorithms:
Sequence alignment can be performed using different algorithms, each with its advantages and disadvantages.
The most common algorithms include pairwise alignment, multiple sequence alignment, and global and local alignment.
Pairwise alignment compares two sequences to identify regions of similarity and difference. The algorithm is based
on the dynamic programming method, which calculates the optimal alignment score by assigning scores to each match,
mismatch, and gap. The Needleman-Wunsch algorithm (Needleman & Wunsch, 1970) and the Smith-Waterman algorithm (Smith & Waterman, 1981)
are two commonly used dynamic programming algorithms (Figure 1).
)
Figure 1. Pairwise sequence alignment in which two sequences are aligned. Types of algnment are highlighted; Local and Global.
Multiple sequence alignment, on the other hand, aligns more than two sequences and allows researchers to identify
conserved regions among them. The algorithm is based on the progressive alignment method, which aligns sequences
pairwise and then combines them into a multiple sequence alignment. The ClustalW and Clustal Omega algorithms are
widely used for multiple sequence alignment (Figure 2).
)
Figure 2. Multiple sequence alignment in which more than a pair of sequences are aligned.
This type of algnment exposes the positions and variations of amino acids or nucliec acids substitution between a number of seqeunces of proteins or genes. This helps to discover conserved regions in different species amongst other important conclusions.
This type of algnment exposes the positions and variations of amino acids or nucliec acids substitution between a number of seqeunces of proteins or genes. This helps to discover conserved regions in different species amongst other important conclusions.
Global alignment aligns the entire sequence, while local alignment aligns only the most similar regions.
The global alignment algorithm is used to compare two sequences with similar lengths and regions of high
similarity. The local alignment algorithm is used to identify similar regions in sequences with different lengths, see also Figure 1.
Percentage and Scoring Matrices Methods:
Scoring matrices are used to calculate the similarity between sequences (Durbin et al., 1998). The most widely used
scoring matrices are the PAM and BLOSUM matrices. The PAM matrix was developed by Margaret Dayhoff in the 1970s and
is based on the evolutionary distance between protein sequences. The BLOSUM matrix, on the other hand, is based on
the observation that highly conserved regions in proteins are more functionally important than less conserved regions.
Percentage identity is a measure of the similarity between two sequences, expressed as a percentage of identical residues
in the aligned sequences. It is calculated by dividing the number of identical residues by the total number of residues
in the aligned sequences (Figure 3).
)
Figure 3. BLAST alignment output showing a number of alignment evaluation terms including identifies and postives percentage and score value based on BLOSUM scoring matrix.
Dot-Matrix Method For Sequence Alignment:
The dot-matrix (also known as the dot plot) is a graphical method for comparing two sequences by plotting matches in a 2D grid. It is not a competitor to alignment algorithms as it's a conceptual and visual precursor to them and is used to show where alignment is possible (Figure 4).

The dot-matrix method represents one of the earliest and most intuitive approaches for comparative sequence analysis, providing a graphical means to identify regions of similarity between two biological sequences. In this method, one sequence is arranged along the horizontal axis and the other along the vertical axis of a matrix, and dots are placed at positions where residues match or satisfy a predefined similarity criterion. Continuous diagonal lines in the resulting plot indicate regions of high similarity, while interruptions or shifts in the diagonal reflect substitutions, insertions, or deletions, thereby offering qualitative insight into evolutionary relationships and structural conservation. Although the dot-matrix approach does not yield an explicit optimal alignment or incorporate scoring schemes such as gap penalties, it remains a valuable exploratory tool for detecting repeats, inversions, and conserved domains, and for guiding subsequent application of more rigorous dynamic programming algorithms. Its conceptual simplicity has also played an important pedagogical role in illustrating the principles underlying modern sequence alignment techniques (Mount, 2004; Durbin et al., 1998).
🕮 Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J.(1990). Basic Local Alignment Search Tool. Journal of molecular biology, 1990 Oct 5;215(3):403-10. doi: 10.1016/S0022-2836(05)80360-2.
🕮 Callaway E. (2020) The race for coronavirus vaccines: a graphical guide. Nature. Oct;586(7830):506-508.
🕮 Dayhoff, M. O., & Eck, R. V. (1966). Atlas of Protein Sequence and Structure, 1967-1968. National Biomedical Research Foundation, Washington, D.C. 5
🕮 De Clercq, E. (2010). The discovery of antiviral agents: ten different compounds, ten different stories. Medical Research Reviews, 30(3), 533-582.
🕮 Dong E, Du H, Gardner L. (2020) An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. May;20(5):533-534. doi: 10.1016/S1473-3099(20)30120-1.
🕮 Durbin R., Eddy, S. R., Krogh, A., & Mitchison, G. (1998) Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press.
🕮 Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. Journal of Molecular Evolution, 1981;17(6):368-76. doi: 10.1007/BF01734359
🕮 Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution, 39(4), 783–791.
🕮 Fitch, W. M. (1971). Toward defining the course of evolution: minimum change for a specific tree topology. Systematic Biology, 20(4), 406–416. 10.1093/sysbio/20.4.406
🕮 Gibrat, J. F., Madej, T., & Bryant, S. H. (1996). Surprising similarities in structure comparison. Current opinion in structural biology, 6(3), 377-385.
🕮 Grubaugh ND, Hanage WP, Rasmussen AL. (2020) Making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear. Cell. Aug 20;182(4):794-795. doi: 10.1016/j.cell.2020.06.040.
🕮 Henikoff, S., & Henikoff, J. G. (1992). Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences, 89(22), 10915-10919.
🕮 Katoh, K., Misawa, K., Kuma, K., & Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30(14), 3059–3066.
🕮 Katoh, K., & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780.
🕮 Kerem, B., Rommens, J. M., Buchanan, J. A., Markiewicz, D., Cox, T. K., Chakravarti, A., ... & Collins, F. S. (1989). Identification of the cystic fibrosis gene: genetic analysis. Science, 245(4922), 1073-1080.
🕮 Margoliash, E. (1963). Primary structure and evolution of cytochrome c. Proceedings of the National Academy of Sciences, 50(4), 672-679.
🕮 Mount, D. W. (2004). Bioinformatics: Sequence and Genome Analysis (2nd ed.). Cold Spring Harbor Laboratory Press.
🕮 Needleman, S. B., & Wunsch, C. D. (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. Journal of molecular biology, 48(3), 443-453.
🕮 Taylor, W. R., & Orengo, C. A. (1989). Protein structure alignment. Journal of molecular biology, 208(1), 1-22.
🕮 Rachedi A. (2020) Mutations in the SARS-CoV-2 complete genome sequences from strains isolated in Blida province, Algeria. JNBGP: Journees Nationales virtuelles de bioinformatique: Genomique et proteomique, October 09-10. doi: 10.13140/RG.2.2.20838.45123.
🕮 Russell, R. B., & Barton, G. J. (1992). Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins: Structure, Function, and Bioinformatics, 14(2), 309-323.
🕮 Saitou, N., & Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution Jul 4(4), 406-425. doi: 10.1093/oxfordjournals.molbev.a040454
🕮 Smith, T. F., & Waterman, M. S. (1981). Identification of common molecular subsequences. Journal of molecular biology, Mar 25;147(1):195-7. doi: 10.1016/0022-2836(81)90087-5.
🕮 Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. (2020) Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. Apr 16;181(2):281-292.e6. doi: 10.1016/j.cell.2020.02.058.
🕮 Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Computer applications in the biosciences: CABIOS, 13(5), 555-556.
🕮 Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. (2020) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. Mar 16;6:14. doi: 10.1038/s41421-020-0153-3.
.<br> <a href=\"\" target=\"_blank\">Link to reference</a>&pth=SAIB&wdr=','related images',1000,800))
Figure 4. Example of a dot plot showing sequence self-similarity. The main diagonal represents identity, while off-diagonal lines indicate repeats or conserved regions (adapted from Wikimedia Commons).
Motives for Biology, Biotechnology, and Disease Fighting:
Sequence alignment has numerous applications in biology, biotechnology, and disease fighting.
In biology, sequence alignment is used to study the evolutionary history of species and identify
conserved regions in proteins that may have functional significance. In biotechnology, sequence
alignment is used to design new drugs, enzymes, and vaccines based on conserved regions in proteins.
In disease fighting, sequence alignment is used to diagnose genetic diseases and identify potential
targets for drug development.
Looking at the recent COVID-19 pandemic, the case of SARS-CoV-2, the virus responsible for the disease, sequence
alignment has played a critical role in both understanding the virus and developing vaccines and antiviral drugs.
Firstly, sequence alignment has helped researchers to identify the genetic makeup of SARS-CoV-2 and compare
it to other related viruses, such as SARS-CoV-1 and MERS-CoV. By aligning the genetic sequences of these viruses,
researchers have been able to identify similarities and differences between them, which has provided insights into
the origins, transmission, and virulence of SARS-CoV-2 (Grubaugh et. al., 2020).
Furthermore, sequence alignment has been used to identify the specific proteins and epitopes (i.e., small protein
fragments that can elicit an immune response) that are most important for developing effective vaccines and antiviral
drugs against SARS-CoV-2 (Walls et. al., 2020). For example, by aligning the sequences of the SARS-CoV-2 spike protein
with those of related viruses, researchers have been able to identify mutations in the regions of the spike protein
that are most likely to elicit an immune response, and these regions have been targeted in the development of
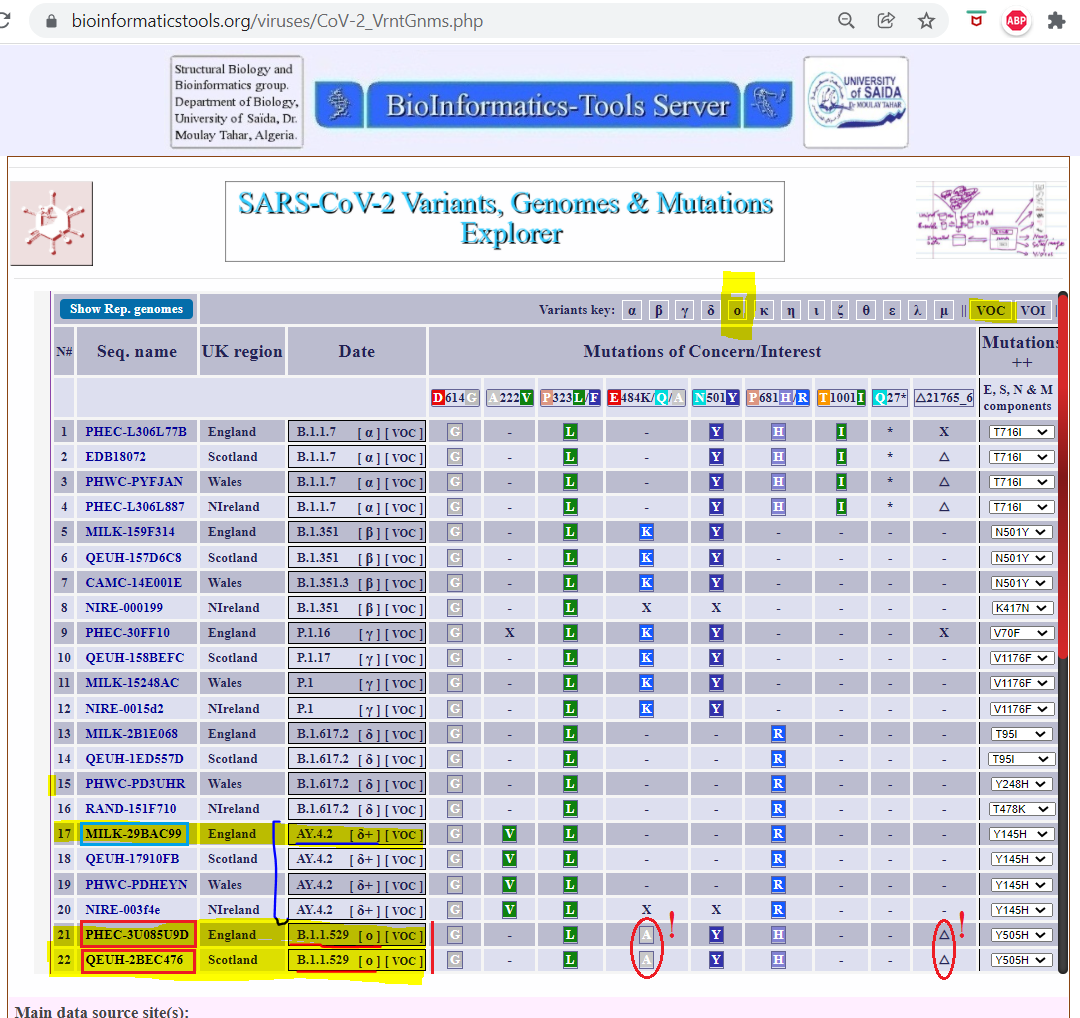
several COVID-19 vaccines (Rachedi, 2020), refer to Figure 5.
)
Figure 5. Table highlighting a number of mutations in the Spike gene that distincted the different variants of the SARS-CoV-2 responsible for the COVID-19 disearse
See: https://bioinformatics.univ-saida.dz/viruses/CoV-2_VrntGnms.php
See: https://bioinformatics.univ-saida.dz/viruses/CoV-2_VrntGnms.php
In addition to vaccine development (Callaway, 2020), sequence alignment has also been used to identify potential drug
targets for SARS-CoV-2 (Zhou et. al., 2020). For example, by aligning the genetic sequences of SARS-CoV-2 with those
of related viruses, researchers have been able to identify conserved regions of the virus that may be vulnerable to
targeting by antiviral drugs. One example is the main protease (Mpro) enzyme, which is essential for the replication of
SARS-CoV-2 and is highly conserved among coronaviruses. By aligning the Mpro sequences from different coronaviruses,
researchers have been able to identify potential drug candidates that can inhibit Mpro and block viral replication.
Phylogenetic Trees:
Sequence alignment plays a fundamental role in the construction of phylogenetic trees, which are graphical representations of the evolutionary relationships among species or groups of organisms. By comparing homologous DNA or protein sequences, researchers can infer patterns of similarity that reflect shared ancestry and evolutionary divergence (Yang, 1997). Importantly, these similarities are interpreted within an algorithmic framework, where sequence data are used as input for computational methods that reconstruct evolutionary histories.
Phylogenetic inference involves analyzing aligned sequences to quantify similarities and differences, typically through models of sequence evolution. These models describe the probabilities of substitutions over time and allow the transformation of sequence alignments into evolutionary distances or likelihood scores. Based on these quantities, various classes of algorithms can be applied to reconstruct tree topologies. Distance-based methods, such as Neighbor-Joining (Saitou & Nei, 1987), construct trees by minimizing overall branch lengths, while clustering approaches like UPGMA assume a constant rate of evolution. In contrast, character-based methods such as Maximum Parsimony (Fitch, 1971) and Maximum Likelihood (Felsenstein, 1981) evaluate alternative tree topologies based on the number of evolutionary changes or the probability of observing the data under a given model. These approaches differ in their assumptions, computational complexity, and accuracy, reflecting the trade-offs inherent in phylogenetic reconstruction.
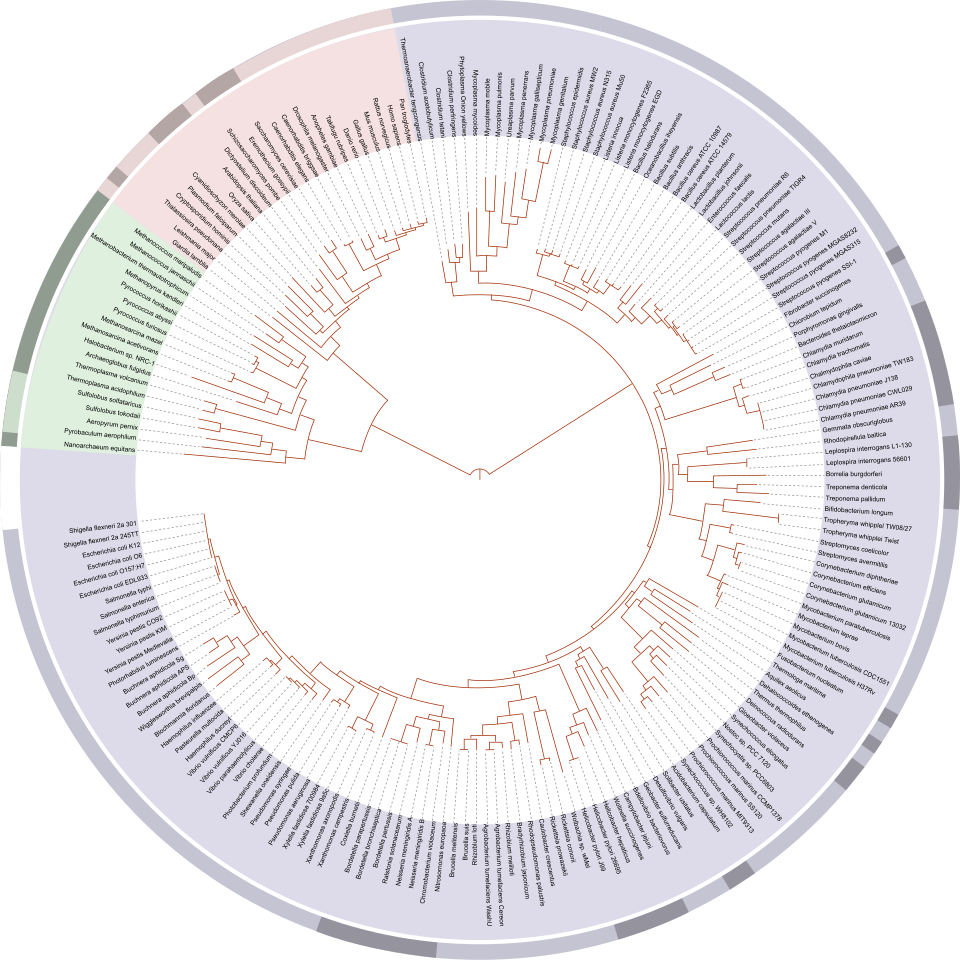
Phylogenetic trees derived from sequence alignments provide critical insights into evolutionary biology. They can be used to infer common ancestry, trace the emergence and distribution of specific traits, and investigate the diversification of lineages over time (Figure 6). Moreover, phylogenetic methods are widely applied in comparative genomics, molecular epidemiology, and pathogen tracking, where they enable the analysis of evolutionary dynamics and transmission patterns.
Bootstrap resampling is a widely used statistical method for assessing the reliability of inferred phylogenetic trees. In this approach, originally introduced by Joseph Felsenstein (Felsenstein, 1985), multiple pseudo-replicate datasets are generated by randomly resampling columns (sites) from the original multiple sequence alignment with replacement. For each replicate, a phylogenetic tree is reconstructed using the same algorithm, and the frequency with which a given clade or branch appears across all replicates is calculated. These frequencies, typically expressed as bootstrap support values (percentages), provide an estimate of the statistical confidence in each branch of the tree. High bootstrap values indicate that a particular grouping is consistently supported by the data, whereas low values suggest uncertainty or insufficient phylogenetic signal. As such, bootstrap analysis serves as an essential complement to phylogenetic reconstruction algorithms, enabling a more robust interpretation of evolutionary relationships.
The integration of sequence alignment with phylogenetic algorithms thus provides a systematic and quantitative framework for studying evolutionary relationships. By combining biological data with computational models and optimization techniques, phylogenetic analysis enhances our understanding of the origin, diversification, and functional evolution of life.
.<br> <a href=\"\" target=\"_blank\">Link to reference</a>&pth=SAIB&wdr=wider','related images',1600,1400))
Figure 6. Example of a phylogenetic tree illustrating evolutionary relationships among major groups of organisms. Branching patterns reflect inferred common ancestry based on molecular sequence comparisons (image adapted from Wikimedia Commons).
Sequence Alignment and Structure Alignment:
Sequence alignment and structure alignment are complementary approaches widely used in bioinformatics to investigate relationships between biological macromolecules. Sequence alignment involves the comparison of nucleotide or amino acid sequences to identify regions of similarity that may indicate homology, shared function, or evolutionary relationships. In contrast, structure alignment compares the three-dimensional conformations of proteins or other macromolecules by superimposing their atomic coordinates to identify spatial similarities and conserved structural features (Russell & Barton, 1992).
From a computational perspective, both sequence and structure alignment can be formulated as optimization problems. Sequence alignment algorithms, such as the Needleman–Wunsch algorithm and Smith–Waterman algorithm, aim to maximize a similarity score based on substitution matrices and gap penalties using dynamic programming techniques. In contrast, structure alignment algorithms seek to minimize the spatial distance between corresponding atoms, typically quantified by the root-mean-square deviation (RMSD), through iterative superposition and geometric optimization. These differing objective functions reflect the distinct nature of sequence and structural data, while highlighting their shared foundation as computational alignment problems.
While sequence alignment focuses on primary structure, structure alignment captures information at the level of tertiary structure, which is generally more conserved during evolution. Consequently, structural comparisons can reveal evolutionary relationships that are not detectable from sequence data alone, particularly among distantly related proteins with low sequence identity but conserved folds.
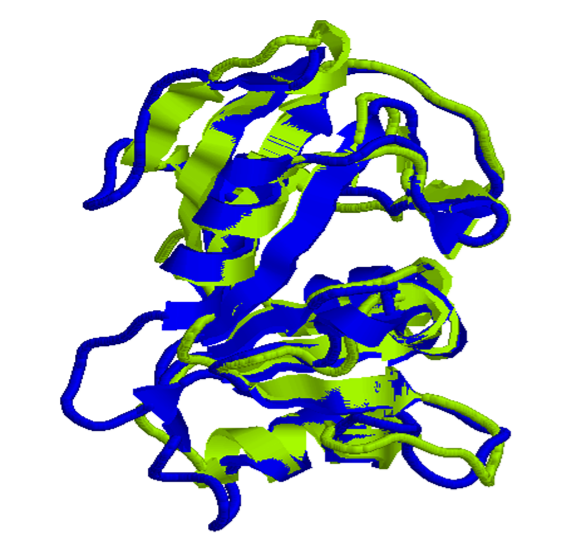
For example, pairwise sequence alignment of the dihydrofolate reductase (DHFR) enzyme from human and Lactobacillus casei shows only ~47.4% sequence similarity. In contrast, structural alignment of their conserved core regions (PDB IDs: 1DHF and 3DFR) yields a root-mean-square deviation (RMSD) of approximately 1.70 Å, indicating a high degree of structural conservation (Figure 7). This example illustrates how protein structure can remain conserved even when sequence similarity is moderate, reflecting the strong evolutionary constraints imposed by functional requirements.
 from human and Lactobacillus casei.
Superposition of the three-dimensional structures of DHFR from human (PDB ID: 1DHF, shown in blue) and L. casei (PDB ID: 3DFR, shown in green). Despite moderate sequence similarity (~47.4%), the proteins exhibit a highly conserved overall fold, with a root-mean-square deviation (RMSD) of approximately 1.70 Å for the aligned core regions. This highlights the greater evolutionary conservation of protein structure relative to sequence and underscores the importance of structural alignment in identifying homologous relationships and preserving functional features.<br> <a href=\"\" target=\"_blank\">Link to reference</a>&pth=SAIB&wdr=','related images',1000,800))
Figure 7. Structural alignment of dihydrofolate reductase (DHFR) from human and Lactobacillus casei.
Superposition of the three-dimensional structures of DHFR from human (PDB ID: 1DHF, shown in blue) and L. casei (PDB ID: 3DFR, shown in green). Despite moderate sequence similarity (~47.4%), the proteins exhibit a highly conserved overall fold, with a root-mean-square deviation (RMSD) of approximately 1.70 Å for the aligned core regions. This highlights the greater evolutionary conservation of protein structure relative to sequence and underscores the importance of structural alignment in identifying homologous relationships and preserving functional features.
Despite these differences, sequence and structure alignment are often used synergistically. Sequence alignment can identify conserved residues that are critical for function or stability, whereas structure alignment can reveal conserved folds, active sites, and binding pockets that may not be apparent at the sequence level. Together, these approaches provide a more comprehensive framework for understanding protein function, evolutionary relationships, and structure–function correlations.
Online Bioinformatics Tools:
Online bioinformatics tools have greatly facilitated access to sequence alignment methods, making advanced computational analyses available to researchers worldwide without the need for local infrastructure. These platforms integrate sophisticated alignment algorithms into user-friendly web interfaces, enabling rapid comparison and analysis of biological sequences.
Among the most widely used tools is BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi), which employs heuristic algorithms to identify regions of local similarity between a query sequence and sequences in large biological databases (Altschul et al., 1990). Its speed and efficiency make it particularly suitable for large-scale database searches, although it does not guarantee globally optimal alignments.
For multiple sequence alignment (MSA), tools such as Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) and MUSCLE (https://www.ebi.ac.uk/Tools/msa/muscle/) are commonly used. These programs rely on progressive and iterative alignment strategies, often guided by distance-based methods and phylogenetic tree construction, to align multiple sequences efficiently. Clustal Omega is optimized for scalability and can handle thousands of sequences, whereas MUSCLE emphasizes high alignment accuracy through iterative refinement.
Other tools, such as MAFFT, further extend multiple sequence alignment methodologies by incorporating fast Fourier transform (FFT)-based strategies to accelerate the detection of homologous regions (Katoh et al., 2002). In MAFFT, sequence similarity is partially evaluated in the frequency domain, allowing rapid identification of conserved motifs and reducing computational complexity compared to traditional dynamic programming approaches. This enables the efficient alignment of large datasets while maintaining high accuracy, particularly when combined with iterative refinement techniques in later stages of the alignment process (Katoh & Standley, 2013).
These tools illustrate the practical implementation of sequence alignment algorithms, balancing computational efficiency and biological accuracy. Their widespread use underscores the importance of algorithmic design in enabling large-scale comparative analyses in modern bioinformatics.
Discussion
Sequence alignment has a wide range of applications in biology and biotechnology, serving as a foundational step in many computational and experimental analyses. In evolutionary biology, alignment algorithms are used to compare homologous sequences across species, enabling the inference of phylogenetic relationships and the reconstruction of evolutionary histories. By identifying conserved and divergent regions within aligned sequences, researchers can detect signatures of selective pressure and functional constraint, providing insight into molecular evolution (Durbin et al., 1998).
Sequence alignment also plays a critical role in mutation detection and medical genomics. By aligning a patient’s genomic or proteomic sequences to a reference sequence, computational algorithms can identify substitutions, insertions, and deletions that may be associated with disease. This approach has been instrumental in the identification of mutations underlying genetic disorders such as cystic fibrosis, where specific variants in the CFTR gene can be detected through sequence comparison. More broadly, alignment-based analyses support the development of diagnostic tools and personalized medicine by enabling the systematic identification and annotation of clinically relevant variants.
In biotechnology and drug discovery, sequence alignment provides a powerful means of identifying and characterizing therapeutic targets. By comparing protein sequences involved in disease pathways, conserved functional domains and active sites can be identified, guiding rational drug design. For example, comparative sequence and structural analyses of viral proteins have been essential in understanding pathogen evolution and host interaction mechanisms. Alignment of genomic sequences of SARS-CoV-2 and its emerging variants has enabled the tracking of mutations affecting transmissibility and immune escape, contributing to vaccine development and optimization. Similarly, studies of the human immunodeficiency virus (HIV) protease enzyme have led to the development of protease inhibitors, a major class of antiretroviral drugs used in HIV therapy (De Clercq, 2010).
Furthermore, sequence alignment is closely linked to structural and functional analysis of proteins. By aligning sequences of proteins with known three-dimensional structures to those of unknown structure, researchers can infer structural features, functional residues, and evolutionary relationships. This principle underlies homology modeling and has been significantly enhanced by recent advances in structure prediction methods, including AlphaFold. Such integrative approaches allow scientists not only to understand protein function but also to engineer novel biomolecules with desired properties, such as enzymes with enhanced catalytic efficiency or stability.
From an algorithmic standpoint, these applications rely on a spectrum of methods ranging from exact dynamic programming approaches to heuristic and probabilistic models. The choice of algorithm depends on the scale of the data and the required balance between accuracy and computational efficiency, highlighting the central role of algorithm design in modern bioinformatics.
Conclusion
Sequence alignment is a fundamental and versatile tool in bioinformatics that enables the systematic comparison and analysis of biological sequences. By identifying similarities and differences between DNA, RNA, and protein sequences, alignment methods provide critical insights into evolutionary relationships, functional conservation, and molecular mechanisms underlying biological processes. Beyond its biological significance, sequence alignment represents a well-defined computational problem that has driven the development of a wide range of algorithms, from exact dynamic programming techniques to fast heuristic and probabilistic methods.
The integration of sequence alignment into bioinformatics pipelines has facilitated major advances in fields such as comparative genomics, disease genomics, structural biology, and drug discovery. As sequencing technologies continue to evolve and generate increasingly large and complex datasets, the importance of efficient and scalable alignment algorithms becomes even more pronounced. Modern tools and approaches, including database search algorithms and machine learning–based methods, are expanding the scope and capabilities of sequence analysis.
In this context, sequence alignment remains a cornerstone of computational biology, bridging raw biological data and meaningful interpretation. Its continued development and application will be essential for advancing our understanding of the structure, function, and evolution of biomolecules, as well as for addressing emerging challenges in biotechnology and medicine.
References
🕮 Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J.(1990). Basic Local Alignment Search Tool. Journal of molecular biology, 1990 Oct 5;215(3):403-10. doi: 10.1016/S0022-2836(05)80360-2.
🕮 Callaway E. (2020) The race for coronavirus vaccines: a graphical guide. Nature. Oct;586(7830):506-508.
🕮 Dayhoff, M. O., & Eck, R. V. (1966). Atlas of Protein Sequence and Structure, 1967-1968. National Biomedical Research Foundation, Washington, D.C. 5
🕮 De Clercq, E. (2010). The discovery of antiviral agents: ten different compounds, ten different stories. Medical Research Reviews, 30(3), 533-582.
🕮 Dong E, Du H, Gardner L. (2020) An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. May;20(5):533-534. doi: 10.1016/S1473-3099(20)30120-1.
🕮 Durbin R., Eddy, S. R., Krogh, A., & Mitchison, G. (1998) Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press.
🕮 Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. Journal of Molecular Evolution, 1981;17(6):368-76. doi: 10.1007/BF01734359
🕮 Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution, 39(4), 783–791.
🕮 Fitch, W. M. (1971). Toward defining the course of evolution: minimum change for a specific tree topology. Systematic Biology, 20(4), 406–416. 10.1093/sysbio/20.4.406
🕮 Gibrat, J. F., Madej, T., & Bryant, S. H. (1996). Surprising similarities in structure comparison. Current opinion in structural biology, 6(3), 377-385.
🕮 Grubaugh ND, Hanage WP, Rasmussen AL. (2020) Making sense of mutation: what D614G means for the COVID-19 pandemic remains unclear. Cell. Aug 20;182(4):794-795. doi: 10.1016/j.cell.2020.06.040.
🕮 Henikoff, S., & Henikoff, J. G. (1992). Amino acid substitution matrices from protein blocks. Proceedings of the National Academy of Sciences, 89(22), 10915-10919.
🕮 Katoh, K., Misawa, K., Kuma, K., & Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30(14), 3059–3066.
🕮 Katoh, K., & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780.
🕮 Kerem, B., Rommens, J. M., Buchanan, J. A., Markiewicz, D., Cox, T. K., Chakravarti, A., ... & Collins, F. S. (1989). Identification of the cystic fibrosis gene: genetic analysis. Science, 245(4922), 1073-1080.
🕮 Margoliash, E. (1963). Primary structure and evolution of cytochrome c. Proceedings of the National Academy of Sciences, 50(4), 672-679.
🕮 Mount, D. W. (2004). Bioinformatics: Sequence and Genome Analysis (2nd ed.). Cold Spring Harbor Laboratory Press.
🕮 Needleman, S. B., & Wunsch, C. D. (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. Journal of molecular biology, 48(3), 443-453.
🕮 Taylor, W. R., & Orengo, C. A. (1989). Protein structure alignment. Journal of molecular biology, 208(1), 1-22.
🕮 Rachedi A. (2020) Mutations in the SARS-CoV-2 complete genome sequences from strains isolated in Blida province, Algeria. JNBGP: Journees Nationales virtuelles de bioinformatique: Genomique et proteomique, October 09-10. doi: 10.13140/RG.2.2.20838.45123.
🕮 Russell, R. B., & Barton, G. J. (1992). Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins: Structure, Function, and Bioinformatics, 14(2), 309-323.
🕮 Saitou, N., & Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution Jul 4(4), 406-425. doi: 10.1093/oxfordjournals.molbev.a040454
🕮 Smith, T. F., & Waterman, M. S. (1981). Identification of common molecular subsequences. Journal of molecular biology, Mar 25;147(1):195-7. doi: 10.1016/0022-2836(81)90087-5.
🕮 Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. (2020) Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. Apr 16;181(2):281-292.e6. doi: 10.1016/j.cell.2020.02.058.
🕮 Yang, Z. (1997). PAML: a program package for phylogenetic analysis by maximum likelihood. Computer applications in the biosciences: CABIOS, 13(5), 555-556.
🕮 Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. (2020) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. Mar 16;6:14. doi: 10.1038/s41421-020-0153-3.